Bất kỳ doanh nghiệp nào muốn tham gia kinh doanh tạp thị trường Hoa Kỳ về thiết bị y tế loại I và II đều phải nộp hồ sơ 510K cho FDA. Trừ trường hợp thiết bị được miễn trừ yêu cầu theo Đạo luật FDA và không vượt qua giới hạn miễn trừ trong các chương trình quy định phân lọi thiết bị. Vậy đăng ký 510K FDA là gì và đăng kí như thế nào.
Đăng ký FDA 510k là gì?
Hồ sơ 510(k) là một hồ sơ nộp trước khi đưa thiết bị ra thị trường nhằm chứng minh rằng thiết bị đó an toàn và hiệu quả tương đương với một thiết bị đã được lưu hành hợp pháp tại Hoa Kỳ (thiết bị “tiền quy định” hoặc thiết bị “tiền chỉnh sửa”).

Người nộp hồ sơ phải so sánh thiết bị của họ với một hoặc nhiều thiết bị đang được lưu hành hợp pháp khác và chứng minh rằng thiết bị của họ là tương đương về an toàn và hiệu quả. Để được coi là tương đương về bản chất, thiết bị mới cần:
Có cùng mục đích sử dụng với thiết bị tham chiếu; và
Có cùng đặc điểm công nghệ hoặc, nếu khác, không đặt ra những câu hỏi an toàn/hiệu quả mới
Dữ liệu gửi đến FDA chứng minh thiết bị mới an toàn và hiệu quả tương đương thiết bị tham chiếu.
Việc tuyên bố tương đương không yêu cầu hai thiết bị phải hoàn toàn giống nhau. FDA sẽ xác nhận sự tương đồng về mục đích sử dụng và đảm bảo rằng bất kỳ khác biệt công nghệ nào cũng không gây ra câu hỏi mới về an toàn hoặc hiệu quả. Sau đó, FDA sẽ đánh giá tính an toàn và hiệu quả thông qua các dữ liệu hiệu suất như thử nghiệm trong phòng thí nghiệm, dữ liệu lâm sàng, thử nghiệm kỹ thuật, kiểm tra độ vô trùng, độ tương thích sinh học, v.v.
Thiết bị không được phép đưa ra thị trường Hoa Kỳ cho đến khi người nộp hồ sơ nhận được thư xác nhận tương đương từ FDA. Nếu thiết bị bị đánh giá là không tương đương, người nộp có thể:
Nộp lại hồ sơ 510(k) với dữ liệu mới;
Yêu cầu phân loại lại thiết bị theo quy trình De Novo (Class I hoặc II);
Gửi đơn đề nghị phân loại lại;
Nộp hồ sơ xin phê duyệt trước khi lưu hành (PMA).
Ai phải nộp hồ sơ 510(k)?
FD&C Act và quy định 21 CFR 807 không quy định cụ thể ai là người phải nộp, mà quy định rõ hành động nào khiến việc nộp hồ sơ là bắt buộc. Bốn nhóm bắt buộc phải nộp:

Nhà sản xuất nội địa đưa thiết bị ra thị trường Hoa Kỳ
Bao gồm các nhà sản xuất thiết bị hoàn chỉnh. Phụ kiện bán cho người dùng cuối cũng được xem là thiết bị hoàn chỉnh.
Nhà sản xuất linh kiện không cần nộp 510(k) trừ khi linh kiện được quảng bá như bộ phận thay thế cho người dùng cuối.
Các nhà gia công theo hợp đồng không cần nộp 510(k) nếu sản xuất theo thông số kỹ thuật của đơn vị khác.

Đơn vị phát triển thông số kỹ thuật (Specification Developers)
Là đơn vị phát triển thiết kế thiết bị và thuê đơn vị khác sản xuất. Chính specification developer là người phải nộp 510(k), không phải nhà sản xuất gia công.

Đơn vị đóng gói/lái nhãn (Repackers hoặc Relabelers)
Phải nộp 510(k) nếu thay đổi nhãn mác (ví dụ: thêm công dụng mới, thay đổi cảnh báo hoặc chống chỉ định…) hoặc nếu hoạt động như tiệt trùng có thể thay đổi điều kiện của thiết bị.

Nhà sản xuất nước ngoài hoặc đại diện tại Mỹ
Giới thiệu sản phẩm vào thị trường Hoa Kỳ. Nếu nhà sản xuất nước ngoài đã có 510(k) hợp lệ thì nhà nhập khẩu không cần nộp lại.
Tất cả các nhà sản xuất thiết bị loại II, III và một số loại I phải tuân thủ thiết kế theo quy định tại 21 CFR 820.30. Hồ sơ thiết kế phải có sẵn để FDA kiểm tra khi thanh tra. Mọi thay đổi trong thiết kế hay sản xuất cần tuân thủ quy định của Hệ thống Chất lượng (21 CFR 820) và có thể yêu cầu nộp lại 510(k).
Những điều kiện doanh nghiệp cần có nếu không muốn đăng ký FDA 510k là gì?

 Doanh nghiệp bán thiết bị chưa hoàn chỉnh cho bên khác gia công tiếp hoặc linh kiện không bán trực tiếp cho người dùng cuối
Doanh nghiệp bán thiết bị chưa hoàn chỉnh cho bên khác gia công tiếp hoặc linh kiện không bán trực tiếp cho người dùng cuối- Thiết bị chỉ đang trong quá trình nghiên cứu/lâm sàng (nhưng vẫn phải tuân IDE 21 CFR 812 nếu thử nghiệm)
- Bạn phân phối thiết bị do hãng nội địa sản xuất và không thay đổi nhãn mác hoặc điều kiện sử dụng
- Bạn chỉ đóng gói/lái nhãn lại nhưng không thay đổi đáng kể nội dung, công dụng hoặc cảnh báo
- Thiết bị đã lưu hành hợp pháp trước ngày 28/5/1976 và chưa thay đổi thiết kế, thành phần, công dụng
- Thiết bị do hãng nước ngoài đã có xác nhận 510(k) và bạn chỉ là bên nhập khẩu
- Thiết bị thuộc danh sách được miễn theo quy định (21 CFR 862–892) và không vượt quá giới hạn miễn (v.d. khác công dụng, công nghệ mới, thiết bị tái sử dụng…)
Quy trình đăng ký FDA 510K là gì
Để nộp hồ sơ 510(k) thành công, cần lên kế hoạch kỹ lưỡng và tuân thủ các yêu cầu pháp lý. Dưới đây là các điểm quan trọng doanh nghiệp cần lưu ý từ giai đoạn đầu:
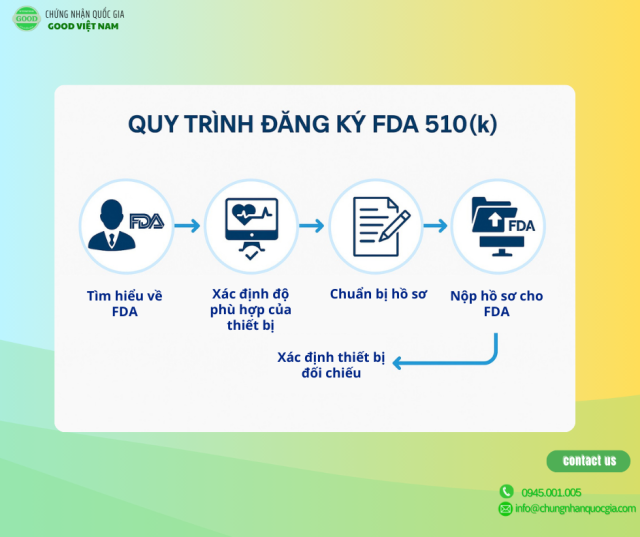

Bước 1: Xác định xem thiết bị y tế của bạn có phù hợp với chương trình FDA 510k hay không
( không phải thiết bị y tế nào cũng phù hợp với chương trình đăng ký FDA 510K. Bạn có thể xác dịnh được thiết bị y tế của mình có phù hợp hay không dựa vào phân loại thiết bị y tế của Hoa Kỳ. Có tất cả 3 level cấp độ phân loại thiết bị y tế:
- Lớp I: Các thiết bị có rủi ro từ thấp đến trung bình, chẳng hạn như băng và que đè lưỡi, thường phải tuân theo các biện pháp kiểm soát chung.
- Lớp II: Các thiết bị có rủi ro trung bình, bao gồm các thiết bị chẩn đoán không xâm lấn và một số dụng cụ phẫu thuật, phải tuân theo các biện pháp kiểm soát đặc biệt và có thể yêu cầu giấy phép 510(k) .
- Lớp III: Các thiết bị có rủi ro cao nhất, chẳng hạn như một số thiết bị cấy ghép, phải trải qua quá trình giám sát theo quy định nghiêm ngặt nhất, đòi hỏi phải có sự chấp thuận trước khi đưa ra thị trường (PMA).

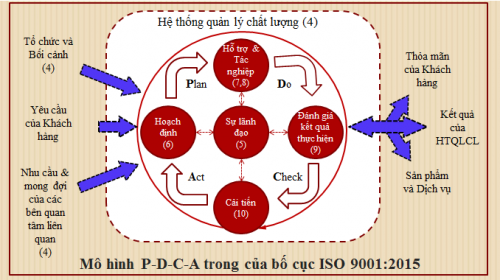
Bước 2: Thiết lập hệ thống QMS
Thiết lập Hệ thống quản lý chất lượng (QMS) mạnh mẽ để đảm bảo chất lượng sản phẩm đồng đều và tuân thủ các quy định của FDA.
sử dụng FDA 820 hoặc ISO 13485 làm yêu cầu cơ bản cho QMS của bạn

Bước 3: Dữ liệu thử nghiệm & lâm sàng
- Xác định nhu cầu về dữ liệu lâm sàng dựa trên mức độ rủi ro của thiết bị của bạn.
- Tiến hành các thử nghiệm và nghiên cứu cần thiết để xác nhận tính an toàn và hiệu suất của thiết bị.
- Đảm bảo rằng các giao thức thử nghiệm được ghi chép đầy đủ và tuân thủ các tiêu chuẩn được công nhận.
- Hãy xem xét những gì có sẵn từ thiết bị tiên đoán.
- Dữ liệu lâm sàng phải được xác thực với bệnh nhân tại Hoa Kỳ!
=> Thực hiện thử nghiệm/đánh giá đầy đủ, đảm bảo dữ liệu hợp lệ tại thị trường Mỹ.

Bước 4: Quản lý rủi ro
Triển khai quy trình quản lý rủi ro mạnh mẽ để xác định, đánh giá và giảm thiểu rủi ro liên quan đến thiết bị của bạn.
Trình bày rõ ràng cách thức tích hợp các biện pháp giảm thiểu rủi ro vào quy trình thiết kế và sản xuất.

Bước 5: Theo dõi các cập nhật mới nhất từ FDA
Thường xuyên kiểm tra các bản cập nhật tài liệu hướng dẫn của FDA có liên quan đến thiết bị của bạn.
Tham dự các hội thảo, hội thảo trực tuyến hoặc buổi đào tạo của FDA để nắm rõ thông tin về các phương pháp hay nhất và kỳ vọng.
VĂN PHÒNG CHỨNG NHẬN QUỐC GIA – GOOD VIỆT NAM
Trụ sở: Số 50B Mai Hắc Đế, P. Nguyên Du, Q. Hai Bà Trưng, Hà Nội
Hotline: 0945.001.005 – 024.2231.5555
E-mail: chungnhanquocgia.com@gmail.com – info@chungnhanquocgia.com
Website: chungnhanquocgia.com
VĂN PHÒNG HÀ NỘI | VĂN PHÒNG ĐÀ NẴNG | VĂN PHÒNG HỒ CHÍ MINH |
Tòa nhà HLT – Số 23 Ngõ 37/2 Dịch vọng, P. Dịch Vọng, Q. Cầu Giấy, Hà Nội | Số 73 Lý Thái Tông, P. Thanh Khê Tây, Q. Thanh Khê, Tp. Đà Nẵng | Tòa nhà PLS, 282 Chu Văn An, Phường 26, Bình Thạnh, TP.HCM |