

Tiêu chuẩn ISO 13485 là tiêu chuẩn về hệ thống quản lý áp dụng trong lĩnh vực sản xuất – kinh doanh dụng cụ, vật tư y tế.
Tiêu chuẩn ISO 13485 nhấn mạnh vào việc hài hoà các yêu cầu của hệ thống quản lý chất lượng với các yêu cầu về luật định đối với ngành thiết bị y tế. Tiêu chuẩn ISO 13458 đã được chấp nhận và được áp dụng rộng rãi cho các nhà sản xuất thiết bị y tế trên toàn thế giới và là một yêu cầu cần phải có trong giai đoạn hiện nay nếu như một tổ chức sản xuất thiết bị y tế muốn sản phẩm của mình được công nhận rộng rãi trên toàn thế giới.
Hiện nay, theo quy định của Pháp luật, việc áp dụng ISO 13485 là một trong những yêu cầu cần phải có đối với các doanh nghiệp kinh doanh, sản xuất trong lĩnh vực trang thiết bị y tế. Cụ thể
Nghị định Chính phủ 36/2016/NĐ_CP ngày 15/5/2016 của Thủ tướng chính phủ về Quản lý trang thiết bị y tế. Này có hiệu lực thi hành từ ngày 01 tháng 7 năm 2016.
“Điều 68. Điều khoản chuyển tiếp
Cơ sở sản xuất trang thiết bị y tế, đã hoạt động trước ngày Nghị định này có hiệu lực thi hành được tiếp tục hoạt động sản xuất nhưng phải hoàn thành việc công bố đủ điều kiện sản xuất trước ngày 01 tháng 7 năm 2017. Riêng đối với quy định về hệ thống quản lý chất lượng: Cơ sở sản xuất trang thiết bị y tế phải hoàn thành việc áp dụng hệ thống quản lý chất lượng ISO 9001 trước ngày 01 tháng 01 năm 2018 và hệ thống quản lý chất lượng ISO 13485 trước ngày ngày 01 tháng 01 năm 2020.”
Như vậy, ngày 01/01/2020 là thời điểm cuối cùng cho các doanh nghiệp thực hiện xây dựng, chứng nhận đạt tiêu chuẩn ISO 13485
![]()
![]()
QUÁ TRÌNH PHÁT TRIỂN CỦA ISO 13485
 ISO là viết tắt tên của tổ chức Quốc tế về tiêu chuẩn hóa (International Organization For Standaraization). Là Tiêu chuẩn về Hệ thống quản lý an toàn cho Sản phẩm y tế nằm trong bộ tiêu chuẩn ISO 13485:2003 (phiên bản năm 2003) do tổ chức ISO ban hành. Trong năm 2011, Ủy ban châu Âu đưa ra một mối quan tâm xung quanh các giả định văn bản quy phạm pháp luật hỗ trợ sự phù hợp với các Chỉ thị thiết bị y tế (Phụ lục ZA, ZB, ZC) trong EN ISO 13485:2003. Kết quả là CEN quyết định xuất bản một ấn bản mới của tiêu chuẩn (EN ISO 13485:2012). Tiêu chuẩn này có một Lời nói đầu được sửa đổi và các Phụ lục ZA, ZB và ZC, nhưng văn bản cốt lõi vẫn không thay đổi.
ISO là viết tắt tên của tổ chức Quốc tế về tiêu chuẩn hóa (International Organization For Standaraization). Là Tiêu chuẩn về Hệ thống quản lý an toàn cho Sản phẩm y tế nằm trong bộ tiêu chuẩn ISO 13485:2003 (phiên bản năm 2003) do tổ chức ISO ban hành. Trong năm 2011, Ủy ban châu Âu đưa ra một mối quan tâm xung quanh các giả định văn bản quy phạm pháp luật hỗ trợ sự phù hợp với các Chỉ thị thiết bị y tế (Phụ lục ZA, ZB, ZC) trong EN ISO 13485:2003. Kết quả là CEN quyết định xuất bản một ấn bản mới của tiêu chuẩn (EN ISO 13485:2012). Tiêu chuẩn này có một Lời nói đầu được sửa đổi và các Phụ lục ZA, ZB và ZC, nhưng văn bản cốt lõi vẫn không thay đổi.
Thiết bị, dụng cụ y tế có vai trò đặc biệt quan trọng trong công tác khám, chữa bệnh và có ảnh hưởng trực tiếp đến đời sống và sức khỏe của con người. Sản phẩm trong lĩnh vực này không chỉ phải đáp ứng các tiêu chuẩn của nhà sản xuất và còn phải tuân thủ các yêu cầu chế định và luật định nhằm đảm bảo thiết bị, dụng cụ y tế cung cấp ra thị trường luôn đáp ứng yêu cầu của khách hàng và các quy định của luật pháp.
ISO 13485 là Tiêu chuẩn quy định các yêu cầu đối với hệ thống quản lý chất lượng áp dụng tại các cơ sở cung cấp dụng cụ y tế và dịch vụ liên quan nhằm đảm bảo khả năng cung cấp sản phẩm đáp ứng yêu cầu của khách hàng và các quy định của luật pháp. Tiêu chuẩn ISO 13485 được xây dựng dựa trên nền tảng của bộ tiêu chuẩn ISO 9000, được tổ chức tiêu chuẩn hóa quốc tế ISO ban hành phiên bản đầu tiên vào tháng 7 năm 2003 (tương đương Tiêu chuẩn quốc gia TCVN ISO 13485:2004).
Mặc dù dựa trên nền tảng bộ tiêu chuẩn ISO 9000, nhưng tiêu chuẩn ISO 13485 nhấn mạnh vào việc hài hoà các yêu cầu của hệ thống quản lý chất lượng với các yêu cầu về luật định đối với ngành thiết bị y tế. Tiêu chuẩn ISO 13485 đã được chấp nhận và được áp dụng rộng rãi cho các nhà sản xuất thiết bị y tế trên toàn thế giới, và là một yêu cầu cần phải có trong giai đoạn hiện nay nếu như một tổ chức sản xuất thiết bị y tế muốn sản phẩm của mình được công nhận rộng rãi trên toàn thế giới.
Là một phần của Hệ thống quản lý chung, vì vậy tổ chức/doanh nghiệp có thể xây dựng hệ thống quản lý chất lượng ISO 13485 độc lập hoặc kết hợp với các hệ thống quản lý khác như ISO 9000, ISO 14000, ISO/IEC 17025, ISO 15189 …
Tổ chức có thể xây dựng hệ thống quản lý chất lượng ISO 13485 độc lập hoặc kết hợp với các hệ thống quản lý khác như ISO 9001, ISO 14001, OHSAS 18001…
PHIÊN BẢN HIỆN HÀNH CỦA ISO 13485
Từ ngày 28 tháng 02 năm 2019, doanh nghiệp phải áp dụng phiên bản mới của Tiêu chuẩn ISO 13485 là ISO 13485 phiên bản ban hành năm 2016 (ISO 13485:2016). Phiên bản này sẽ thay thế phiên bản ISO 13485:2003
Điều đó cũng có nghĩa là việc cập nhật lên phiên bản mới từ phiên bản cũ ISO 13485:2003 và tiêu chuẩn Châu Âu liên quan: EN ISO 13485:2012 cần được thực hiện chậm nhất là vào ngày này.
Phiên bản ISO 13485:2016 đề cao tầm quan trọng của việc cải tiến liên tục được thay thế bằng việc đáp ứng các yêu cầu luật định và các yêu cầu cụ thể của khách hàng, quản lý rủi ro, và duy trì các quá trình hiệu quả, cụ thể là thiết kế, sản xuất và phân phối một cách an toàn các dụng cụ y khoa.
Phiên bản hiện hành của tiêu chuẩn này là ISO 13485:2016 lấy ISO 9001 làm nền tảng, tầm quan trọng của việc cải tiến liên tục được thay thế bằng việc đáp ứng các yêu cầu luật định và các yêu cầu cụ thể của khách hàng, quản lý rủi ro, và duy trì các quá trình sản xuất hiệu quả, các yêu cầu về kiểm soát nhiễm bẩn, vệ sinh sản phẩm, yêu cầu đặc biệt với thiết bị y tế vô trùng và các yêu cầu về truy xuất sản phẩm.
Áp dụng ISO 13485:2016 sẽ đáp ứng được yêu cầu bắt buộc của Nghị định 36/2016/NĐ-CP về quản lý trang thiết bị y tế trong đó yêu cầu Cơ sở sản xuất trang thiết bị y tế phải hoàn thành việc áp dụng hệ thống quản lý chất lượng theo ISO 13485:2016.
Khi Tổ chức được Đánh giá và Cấp giấy chứng nhận ISO 13485:2016, thì các Thủ tục đối với Cơ quan quản lý nhà nước sẽ thuận lợi,dễ dàng hơn, hồ sơ đơn giản hơn khi làm các thủ tục công bố công bố đủ điều kiện sản xuất trang thiết bị y tế, công bố tiêu chuẩn chất lượng, phân loại trang thiết bị y tế, đăng ký lưu hành trang thiết bị y tế.
CÁC YÊU CẦU LUẬT ĐỊNH LIÊN QUAN TỚI ISO 13485
Nghị định Chính phủ 36/2016/NĐ_CP ngày 15/5/2016 của Thủ tướng chính phủ về Quản lý trang thiết bị y tế. Này có hiệu lực thi hành từ ngày 01 tháng 7 năm 2016.
“Điều 68. Điều khoản chuyển tiếp
Cơ sở sản xuất trang thiết bị y tế, đã hoạt động trước ngày Nghị định này có hiệu lực thi hành được tiếp tục hoạt động sản xuất nhưng phải hoàn thành việc công bố đủ điều kiện sản xuất trước ngày 01 tháng 7 năm 2017. Riêng đối với quy định về hệ thống quản lý chất lượng: Cơ sở sản xuất trang thiết bị y tế phải hoàn thành việc áp dụng hệ thống quản lý chất lượng ISO 9001 trước ngày 01 tháng 01 năm 2018 và hệ thống quản lý chất lượng ISO 13485 trước ngày ngày 01 tháng 01 năm 2020.”
Một số nội dung bao gồm:
1. Bố trí nhà xưởng phù hợp yêu cầu vệ sinh, chống nhiễm chéo, nhiễm bẩn
2. Xin giấy phép đủ điều kiện sản xuất
3. Lựa chọn người phụ trách chuyên môn đủ năng lực (theo quy định) trong quản lý sản xuất
4. Phân loại sản phẩm theo nhóm (A, B, C, D)
5. Kiểm định/ thử nghiệm ban đầu những sản phẩm thuộc nhóm B, C, D
6. Xin cấp số lưu hành sản phẩm
7. Xin cấp số lưu hành tự do CFS đối với sản phẩm xuất khẩu
8. Xin giấy xác nhận an toàn cho những sản phẩm đã lưu hành hợp pháp
9. Xin thử nghiệm lâm sàng đối với trang thiết bị thử nghiệm lâm sàng đã hoàn thành thiết kế kiểm định
10. Công bố hợp chuẩn hợp quy trước khi lưu hành sản phẩm
11. Báo cáo định kỳ trước 31/1 hàng năm

CẤU TRÚC CỦA TIÊU CHUẨN ISO 13485:2016
Cấu trúc ISO 13485 được chia thành tám phần, bởi vì nó phù hợp với tiêu chuẩn ISO 9001. Ba phần đầu tiên là phần giới thiệu, với năm phần cuối có chứa các yêu cầu cho Hệ thống quản lý chất lượng. Cụ thể:
1 Phạm vi áp dụng
2 Tài liệu viện dẫn
3 Thuật ngữ và định nghĩa
4 Hệ thống quản lý chất lượng
4.1 Yêu cầu chung
4.2 Yêu cầu về hệ thống tài liệu
5 Trách nhiệm của lãnh đạo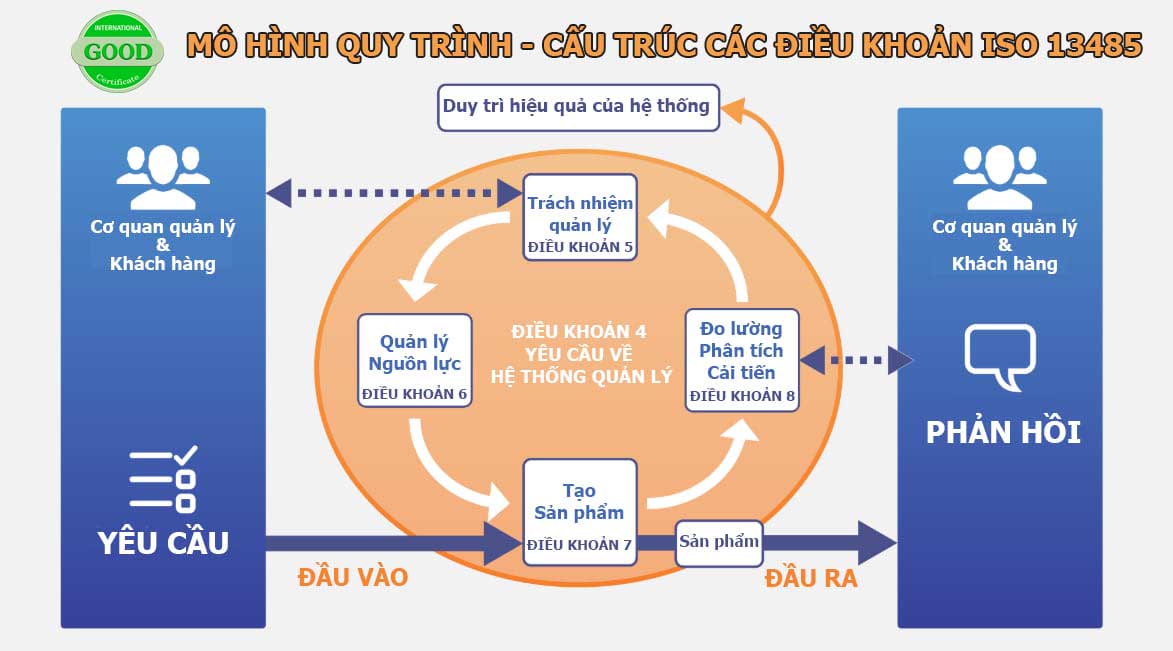
5.1 Cam kết của lãnh đạo
5.2 Hướng vào khách hàng
5.3 Chính sách chất lượng
5.4 Hoạch định
5.5 Trách nhiệm, quyền hạn và trao đổi thông tin
5.6 Xem xét của lãnh đạo
6 Quản lý nguồn lực
6.1 Cung cấp nguồn lực
6.2 Nguồn nhân lực
6.3 Cơ sở hạ tầng
6.4 Môi trường làm việc và kiểm soát nhiễm bẩn
7 Tạo sản phẩm
7.1 Hoạch định việc tạo sản phẩm
7.2 Các quá trình liên quan đến khách hàng
7.3 Thiết kế và phát triển
7.4 Mua hàng
7.5 Sản xuất và cung cấp dịch vụ
7.6 Kiểm soát phương tiện theo dõi và đo lường
8 Đo lường, phân tích và cải tiến
8.1 Khái quát
8.2 Theo dõi và đo lường
8.3 Kiểm soát sản phẩm không phù hợp
8.4 Phân tích các dữ liệu
8.5 Cải tiến
NHỮNG NỘI DUNG CƠ BẢN VÀ ĐIỂM THAY ĐỔI CỦA ISO 13485:2016
Phần từ Mục 4 tới Mục 8 là những yêu cầu của tiêu chuẩn ISO 13485:2016. Nội dung cơ bản của 5 phần này bao gồm:
Phần 4: Hệ thống quản lý chất lượng
Phần này nói về các yêu cầu QMS chung, cũng như các yêu cầu về tài liệu của tiêu chuẩn. Nó bao gồm các yêu cầu về Sổ tay Chất lượng, Kiểm soát Tài liệu và Kiểm soát Hồ sơ, tất cả đều là các tài liệu cần thiết trong QMS.
Thay đổi cơ bản:
Tất cả các quy trình là một phần của hệ thống quản lý chất lượng từ nay cần được tích hợp phương pháp tiếp cận dựa trên rủi ro ngoài quá trình tạo sản phẩm. Rủi ro được xem xét trong phạm vi an toàn và hiệu quả của thiết bị y tế và việc đáp ứng các yêu cầu pháp lý.
Ngoài ra bất kỳ phần mềm nào được sử dụng như một phần của hệ thống quản lý chất lượng phải được xác nhận và ghi lại. Đối với từng thiết bị sản xuất cũng cần được kèm theo các mô tả thiết bị, các thông số và hồ sơ liên quan.
Phần 5: Trách nhiệm quản lý
Các yêu cầu về trách nhiệm quản lý bao gồm nhu cầu quản lý hàng đầu là công cụ trong việc thực hiện và duy trì QMS. Cùng với việc lập kế hoạch cho QMS, cần có sự quản lý hàng đầu để tham gia vào việc xem xét liên tục hệ thống để đảm bảo sự hài lòng và cải thiện của khách hàng.
Thay đổi cơ bản:
Những thay đổi trong phần này chủ yếu liên quan đến việc làm rõ các yêu cầu hiện có liên quan đến quy hoạch hệ thống quản lý chất lượng, trách nhiệm và thẩm quyền, đại diện quản lý và đánh giá quản lý. Điều này giúp hài hòa yêu cầu về kiểm tra xác nhận các phần mềm ứng dụng khác nhau như quản lý chất lượng, kiểm soát quá trình, phần mềm giám sát và đo lường trong các điều khoản khác nhau của bộ tiêu chuẩn
Phần 6: Quản lý tài nguyên
Phần quản lý tài nguyên ngắn gọn, nhưng bao gồm sự cần thiết phải kiểm soát tất cả các tài nguyên, bao gồm nguồn nhân lực, các tòa nhà và cơ sở hạ tầng và môi trường làm việc.
Thay đổi cơ bản:
Các nhà sản xuất thiết bị sẽ có nghĩa vụ xác định các kỹ năng và kinh nghiệm cụ thể cần thiết cho các nhân viên tham gia vào việc duy trì hệ thống quản lý chất lượng. Các tổ chức được chứng nhận ISO 13485 cũng sẽ phải duy trì các hệ thống để đảm bảo rằng nhân viên được duy trì kiến thức thông qua việc đào tạo liên tục và đánh giá tính hiệu quả từ việc đào tạo.
Điều khoản mới trong phần này cũng đề cập đến các vấn đề kiểm soát ô nhiễm cho các thiết bị y tế vô trùng và bao gồm các yêu cầu liên quan đến việc xác nhận các quy trình nhằm đảm bảo tính toàn vẹn và hiệu quả của sản xuất thiết bị vô trùng.
Phần 7: Thực hiện sản phẩm
Các yêu cầu sản phẩm đối phó với tất cả các khía cạnh của việc lập kế hoạch và tạo ra sản phẩm hoặc dịch vụ. Phần này bao gồm các yêu cầu về lập kế hoạch, đánh giá yêu cầu sản phẩm, thiết kế, mua, tạo ra sản phẩm hoặc dịch vụ và kiểm soát thiết bị được sử dụng để giám sát và đo lường sản phẩm hoặc dịch vụ. ISO 9001 cho phép các yêu cầu trong phần bị loại trừ nếu chúng không áp dụng cho công ty (chẳng hạn như một công ty không thiết kế sản phẩm hoặc dịch vụ).
Thay đổi cơ bản:
Khoản 7 hướng tới các yêu cầu cụ thể trong từng lĩnh vực được xác định trong phạm vi mở rộng việc thực hiện sản phẩm. Các nhà sản xuất thiết bị y tế dự kiến sẽ kết hợp các nguyên tắc quản lý rủi ro trong việc xác định việc áp dụng các yêu cầu này.
Phần 8: Đo lường, Phân tích và Cải tiến
Phần cuối cùng này bao gồm các yêu cầu cần thiết để đảm bảo rằng bạn có thể theo dõi xem QMS của bạn có hoạt động tốt hay không. Nó bao gồm đánh giá sự hài lòng của khách hàng, kiểm toán nội bộ, giám sát sản phẩm và quy trình, xử lý sản phẩm không phù hợp, và các hành động khắc phục và phòng ngừa.
Thay đổi cơ bản:
Các nhà sản xuất thiết bị sẽ chuẩn hóa quy trình để thu thập phản hồi từ hoạt động sản xuất, hậu sản xuất và phát triển phương pháp báo hiệu để kết hợp phản hồi vào chương trình quản lý rủi ro. Các yêu cầu liên quan đến việc kiểm tra và kiểm soát các sản phẩm không phù hợp, các yêu cầu liên quan đến các hành động khắc phục và phòng ngừa đã được tăng cường. Các điều khoản mới này đã được tạo ra trong việc giám sát và đo lường để xử lý khiếu nại và báo cáo cho cơ quan quản lý. Hoạt động giúp hoạch định và văn bản hóa các hành động khắc phục và phòng ngừa; và thực hiện những hành động khắc phục trong thời gian sớm nhất.
Các phần này dựa trên chu kỳ Plan-Do-Act-Check, sử dụng các yếu tố này để thực hiện thay đổi trong các quy trình của tổ chức nhằm thúc đẩy và duy trì các cải tiến trong các quy trình.
QUÁ TRÌNH TRIỂN KHAI ÁP DỤNG ISO 13485:2016
Khách hàng có thể tham khảo Kế hoạch tư vấn sau để triển khai áp dụng ISO 13485:2016
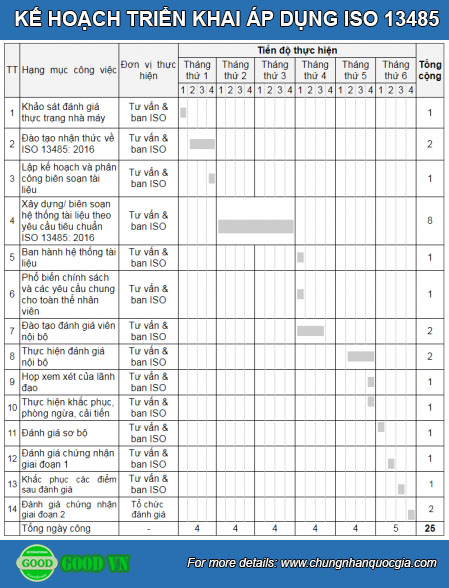
 Dưới đây là các hồ sơ và các quy trình theo ISO 13485 là cần thiết để tuân thủ ISO 13485: 2016.
Dưới đây là các hồ sơ và các quy trình theo ISO 13485 là cần thiết để tuân thủ ISO 13485: 2016.
(Xin lưu ý rằng một số tài liệu sẽ không bắt buộc nếu công ty không thực hiện các quá trình có liên quan):
Vai trò được thực hiện bởi tổ chức theo các yêu cầu quy định pháp luật hiện hành (mục 4.1.1)
Thủ tục và hồ sơ xác nhận giá trị sử dụng việc áp dụng phần mềm máy tính (mục 4.1.6)
Sổ tay Chất lượng (mục 4.2.2)
Hồ sơ thiết bị y tế (mục 4.2.3)
Thủ tục kiểm soát tài liệu (mục 4.2.4)
Thủ tục kiểm soát hồ sơ (mục 4.2.5)
Chính sách chất lượng (khoản 5.3)
Mục tiêu chất lượng (mục 5.4.1)
Trách nhiệm và quyền hạn (mục 5.5.1)
Thủ tục và hồ sơ để xem xét của lãnh đạo (mục 5.6.1)
Thủ tục đào tạo (mục 6.2)
Yêu cầu về cơ sở hạ tầng và các hoạt động bảo trì (mục 6.3)
Yêu cầu đối với môi trường làm việc (mục 6.4.1)
Sắp xếp để kiểm soát các sản phẩm bị ô nhiễm hoặc có khả năng bị ô nhiễm (mục 6.4.2)
Quy trình quản lý rủi ro trong quá trình thực hiện sản phẩm (khoản 7.1)
Đầu ra của kế hoạch thực hiện sản phẩm (khoản 7.1)
Hồ sơ của kết quả xem xét yêu cầu của khách hàng và hành động phát sinh từ nó (mục 7.2.2)
Hồ sơ liên lạc với khách hàng (mục 7.2.3)
Thủ tục thiết kế và phát triển (mục 7.3.1)
Kế hoạch thiết kế và phát triển (mục 7.3.2)
Đầu ra thiết kế và phát triển (mục 7.3.4)
Hồ sơ xem xét thiết kế và phát triển (mục 7.3.5)
Kế hoạch, kết quả và kết luận về kiểm tra xác nhận thiết kế (mục 7.3.6)
Thủ tục chuyển giao kết quả thiết kế và phát triển sản xuất (mục 7.3.8)
Quy trình và hồ sơ kiểm soát sự thay đổi thiết kế và phát triển (mục 7.3.9)
Hồ sơ thiết kế và phát triển (mục 7.3.10)
Thủ tục mua hàng (mục 7.4.1)
Các tiêu chí và hồ sơ đánh giá và lựa chọn nhà cung cấp (mục 7.4.1)
Bản kiểm tra sản phẩm đã mua (mục 7.4.3)
Hồ sơ truy xuất nguồn gốc cho mỗi thiết bị y tế hoặc lô hàng (mục 7.5.1)
Yêu cầu về độ sạch của sản phẩm (mục 7.5.2)
Yêu cầu về tiêu chuẩn lắp đặt và chấp nhận để xác minh việc lắp đặt thiết bị y tế (mục 7.5.3)
Hồ sơ lắp đặt thiết bị y tế và thẩm định lắp đặt (mục 7.5.3)
Thủ tục và hồ sơ dịch vụ thiết bị y tế (mục 7.5.4)
Hồ sơ về quá trình khử trùng (mục 7.5.5)
Thủ tục và hồ sơ xác nhận quá trình sản xuất và cung cấp dịch vụ (mục 7.5.6)
Quy trình và hồ sơ xác nhận quá trình khử trùng và các hệ thống rào vô trùng (mục 7.5.7)
Thủ tục nhận dạng sản phẩm (mục 7.5.8)
Thủ tục truy xuất nguồn gốc (mục 7.5.9.1)
Hồ sơ truy xuất nguồn gốc, tên và địa chỉ của bên nhận vận chuyển (khoản 7.5.9.2)
Báo cáo về những thay đổi về tài sản của khách hàng (mục 7.5.10)
Thủ tục đảm bảo sự phù hợp của sản phẩm (mục 7.5.11)
Thủ tục theo dõi và đo lường (mục 7.6)
Biên bản hiệu chuẩn (mục 7.6)
Thủ tục và hồ sơ xác nhận việc áp dụng phần mềm máy tính dùng để giám sát và đo lường (mục 7.6)
Thủ tục phản hồi của khách hàng (mục 8.2.1)
Thủ tục và hồ sơ xử lý khiếu nại (mục 8.2.2)
Hồ sơ báo cáo cho cơ quan quản lý (mục 8.2.3)
Thủ tục đánh giá nội bộ (mục 8.2.4)
Biên bản đánh giá nội bộ và kết quả (mục 8.2.4)
Thông tin nhận dạng của người có thẩm quyền thông qua sản phẩm (mục 8.2.6)
Thủ tục và hồ sơ kiểm soát sản phẩm không phù hợp (mục 8.3.1)
Biên bản làm lại (khoản 8.3.4)
Thủ tục và hồ sơ để phân tích số liệu (mục 8.4)
Thủ tục và hồ sơ cho hành động khắc phục (khoản 8.5.2)
Thủ tục và hồ sơ về hành động phòng ngừa (mục 8.5.3)
Ngoài các quy trình iso 13485 cần thiết, bạn cần phải áp dụng các quy trình này trong vòng 3 tháng trước khi đánh giá chứng nhận bởi Tổ chức chứng nhận
Mọi nhu cầu hay thắc mắc liên quan đến ISO 13485 .Quý khách hãy liên hệ với chúng tôi để được hỗ trợ tư vấn miễn phí trên mọi tỉnh thành.
GỌI NGAY: 0945.001.005 – 0963.831.555 – 02466.82.0505
CHÚNG TÔI Ở ĐÂY ĐỂ PHỤC VỤ BẠN !




Pingback: các tiêu chuẩn chất lượng - taytou
Pingback: các tiêu chuẩn en - taytou